Home
![]()
Introduction
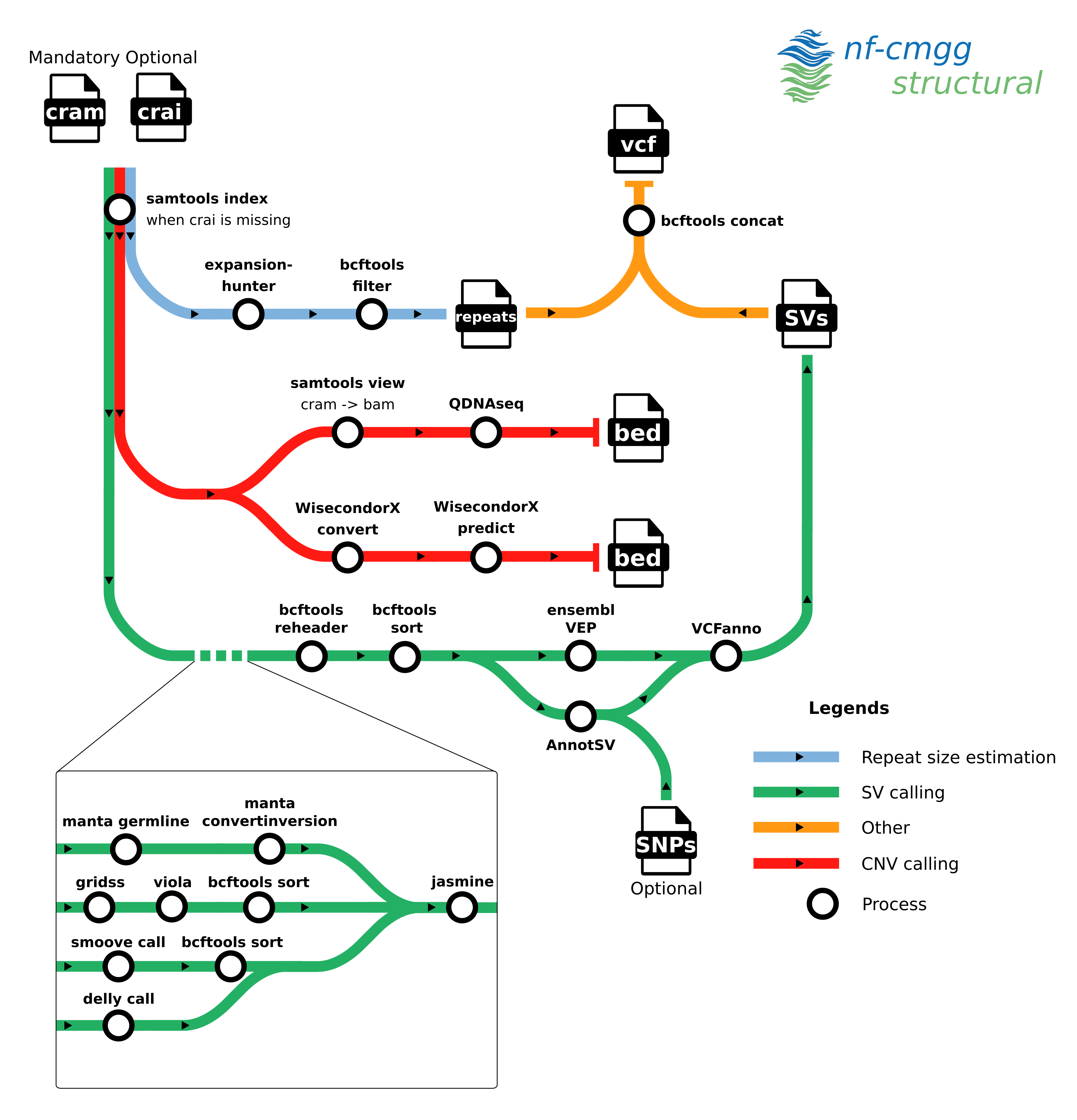
nf-cmgg/structural is a bioinformatics best-practice analysis pipeline for calling structural variants (SVs), copy number variants (CNVs) and repeat region expansions (RREs) from short DNA reads. The pipeline handles the calling of the variants and postprocessing (filtering, annotating...)
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where possible, these processes have been submitted to and installed from nf-core/modules in order to make them available to all Nextflow pipelines!
Usage
Note
If you are new to Nextflow and nf-core, please refer to this page on how
to set-up Nextflow. Make sure to test your setup
with -profile test before running the workflow on actual data.
Now, you can run the pipeline using:
nextflow run nf-cmgg/structural \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Warning
Please provide pipeline parameters via the CLI or Nextflow -params-file option. Custom config files including those
provided by the -c Nextflow option can be used to provide any configuration except for parameters;
see docs.
Documentation
The nf-cmgg/structural pipeline comes with documentation about the pipeline usage and output.
Credits
nf-cmgg/structural was originally written by Nicolas Vannieuwkerke and Mattias Van Heetvelde.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
Citations
An extensive list of references for the tools used by the pipeline can be found in the Citations section.